
大家都在搜

手机验证

询价列表

暂时没有已询价产品
2014年4月
http://molcalx.com/ligandscout
英语
企业
是
无,在线下载
否
药物设计软件LigandScout
3.1
200MB
奥地利Inteligand公司
Windows, Linux, Mac OS X
序列号激活
充足
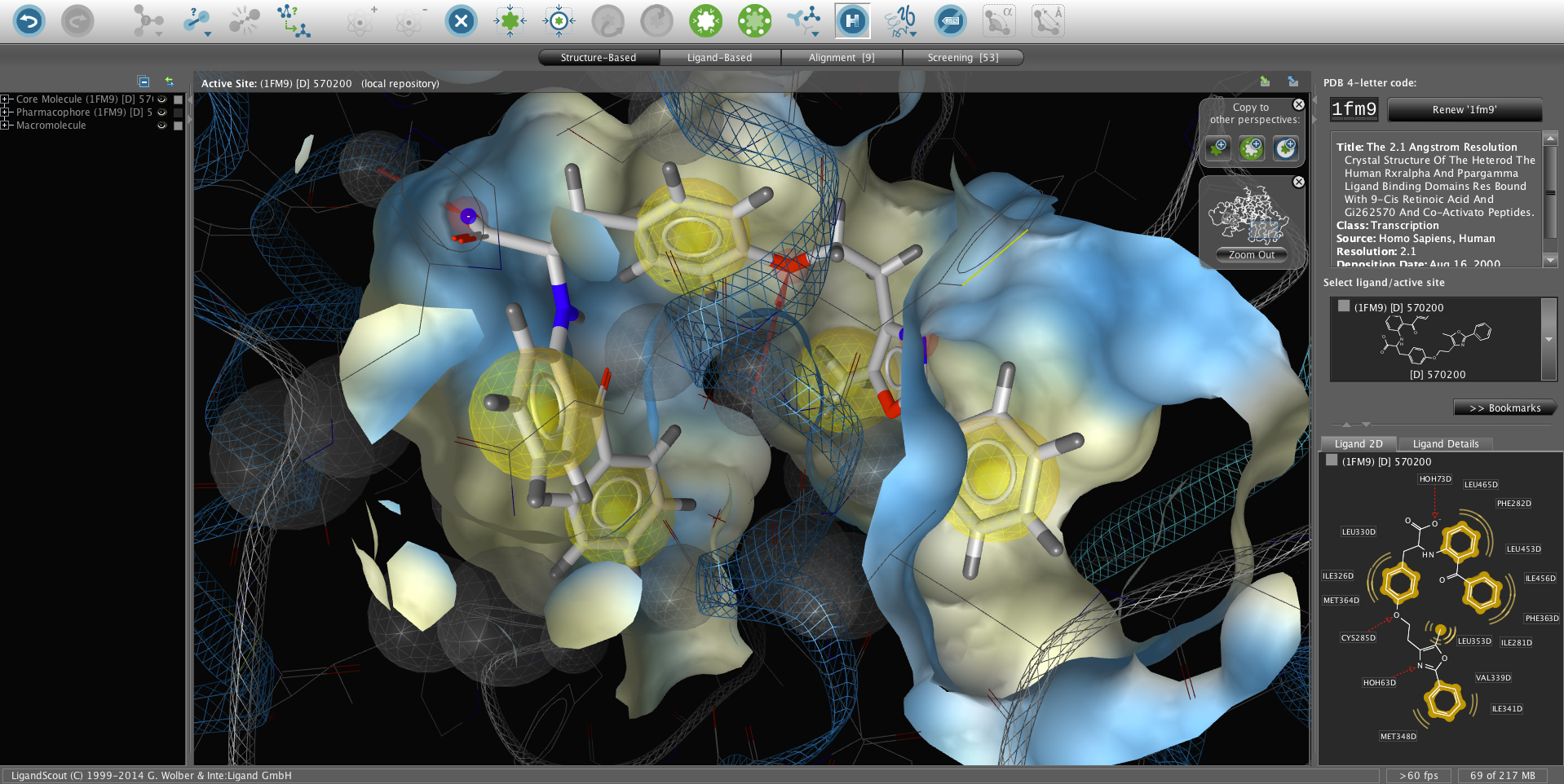
本软件的算法已经在科学杂志上公开发表[1-4]并以我们多年在药效团建模的经验为基础开发了最为先进的药效团应用软 件。LigandScout支持多种常用的药效团格式,可以导出模型给Catalyst (Discovery Studio)、MOE与Phase (Schrodinger)等软件进行虚拟筛选以保证最大的协同工作能力。功能齐全的3D图形用户界面支持多层级撤销(undo),使得复杂地活性位点分 析与药效团建模变得高效、简单明了。我们设计了结合位点分析、基于药效团的叠合以及共有药效团特征的创建等应用使得LigandScout成为基于结构设 计与虚拟筛选的必备工具。Ligandscout还支持所有常见的操作系统。
二、免费测试
您可以下载一个全功能的版本,注册后可以获得一个月的测试授权。如果您需要更长时间的测试或预定一个测试时间、或想获取更多信息或有技术问题要咨询,请。
请您附上您的信息:
姓名:
头衔:如果是学生,请附上您指导老师的信息
单位:比如中山大学
单位地址:用街道名称,比如广州市天河区龙口东路38号2002室
所在部门:比如药学院
Email:
电话:
请将您的信息发送到info@molcalx.com, 我们确认了您的信息之后会给您发软件序列号。
三、算法文献
[1] Wolber, G.; Dornhofer, A. A.; Langer, T.; Efficient overlay of small organic molecules using 3D pharmacophores J. Comput. Aided Mol. Des.; 2007; 20(12); 773-788. DOI: 10.1007/s10822-006-9078-7
[2] Wolber, G.; Langer, T.; LigandScout: 3-D Pharmacophores Derived from Protein-Bound Ligands and Their Use as Virtual Screening Filters. J. Chem. Inf. Model; 2005; 45(1); 160-169. DOI: 10.1021/ci049885e (request reprint)
[3] Wolber, G.; Kirchmair H.; Langer, T.; Structure-Based 3D Pharmacophores: An Alternative to Docking? Plenary talk at the 7th International Conference on Chemical Structures in Noordwijkerhout, 2005
点击下载PDF文件: [2.8 MB]
[4] Wolber G., Dornhofer A. A., Langer T.; Efficient Overlay of Molecular 3D Pharmacophores. Oral presentation at the spring ACS meeting in Atlanta, GA, USA, 2006
点击下载PDF文件: [8.8 MB]
四、主要特性
基于结构的3D药效团设计
精确、快速的虚拟筛选
基于配体的药效团设计
需要更多信息请联系我们
五、系统要求
内存:至少2GB的内存,推荐4GB或以上
操作系统:Windows XP,Windows Vista,MacOS X Leopard (10.5),Sonw Leopard (10.6)或更高的版本,需要安装JRE (Java runtime enviroment).
显卡:支持OpenGL 1.2的硬件加速3D显卡(推荐NVIDIA或ATI)
六、部分国内用户文献
(1)Wang, D.; Zhu, X.; Cui, C.; Dong, M.; Jiang, H.; Li, Z.; Liu, Z.; Zhu, W.; Wang, J.-G. Discovery of Novel Acetohydroxyacid Synthase Inhibitors as Active Agents Against Mycobacterium Tuberculosis by Virtual Screening and Bioassay. J Chem Inf Model 2013. http://dx.doi.org/10.1021/ci3004545
Acetohydroxyacid synthase (AHAS) has been regarded as a promising drug target against Mycobacterium tuberculosis (MTB) as it catalyzes the biosynthesis of branched-chain amino acids. In this study, 23 novel AHAS inhibitors were identified through molecular docking followed by similarity search. The determined IC(50) values range from 0.385 ± 0.026 μM to >200 μM against bacterium AHAS. Five of the identified compounds show significant in vitro activity against H37Rv strains (MICs in the range of 2.5-80 mg/L) and clinical MTB strains, including MDR and XDR isolates. More impressively, compounds 5 and 7 can enhance the killing ability against macrophages infected pathogen remarkably. This study suggests our discovered inhibitors can be further developed as novel anti-MTB therapeutics targeting AHAS.
(2)Yang, H.; Shen, Y.; Chen, J.; Jiang, Q.; Leng, Y.; Shen, J. Structure-Based Virtual Screening for Identification of Novel 11β-HSD1 Inhibitors. Eur J Med Chem 2009, 44, 1167–1171.
Structure-based pharmacophore models were built by using LigandScout and used for virtual screening of the SPECS database to identify new potential 11β-HSD1 inhibitors. As a refinement of the results obtained from virtual 3D pharmacophore screening, the best fitting virtual hits were subjected to docking study. The resulting compds. were tested in an enzyme assay and revealed several compds. with novel scaffolds that show sub-micromolar activity and high selectivity for 11β-HSD1 against 11β-HSD2.
(3)Lu, X.; Chen, Y.; You, Q. Pharmacophore Guided 3D-QSAR CoMFA Analysis of Amino Substituted Nitrogen Heterocycle Ureas as KDR Inhibitors. QSAR Comb. Sci. 2009, 28, 1524–1536.
Vascular endothelial growth factor-2 receptors (VEGR-2) or kinase insertdomain receptor (KDR) is a promising target for the development of novel anticancer drugs. To understand the structural basis for KDR inhibitory activity, 3D-QSAR study using CoMFA anal. was performed on a set of amino substituted nitrogen heterocyclic urea derivs. Due to the flexibility and structural diversity of investigated derivs., we applied pharmacophore based alignment to construct reliable 3D-QSAR models. The pharmacophore model was generated based on the verified docked conformation of compd. 2 with KDR crystal structure using LigandScout 2.0 software. The constructed CoMFA model produced reasonable statistics, with rcv2 =.507 and conventional r2 = 0.982. The predictive power of the developed model was obtained using a test set of 16 mols., giving predictive correlation coeff. of 0.540. Mol. modeling and CoMFA contour anal. were performed to obtain useful information about the structural requirements for the KDR inhibitors which could be utilized in its future design.
(4) Lu, X.-Y.; Chen, Y.-D.; You, Q.-D. 3D-QSAR Studies of Arylcarboxamides with Inhibitory Activity on InhA Using Pharmacophore-Based Alignment. Chemical biology & drug design 2010, 75, 195–203.
Enoyl acyl carrier protein reductase (InhA) is a promising target for the development of antituberculosis drugs. The InhA-bound conformation of an indole-5-amide inhibitor (Genz 10850) (PDB code: IP44) was used to build a pharmacophore model by LigandScout. This model was then successfully used to identify the bioactive conformation and align 40 structurally diverse arylcarboxamide derivatives. Comparative molecular field analysis (CoMFA) and comparative molecular similarity indices analysis (CoMSIA) were performed on arylcarboxamides-based InhA inhibitors based on pharmacophore alignment. The best prediction was obtained with CoMSIA model combining steric and electrostatic fields (, r(2) = 0.972). The model was validated by an external test set, which gave a good predictive value (). Graphical interpretation of the results revealed important structural features of the zarylcarboxamides related to the active site of InhA. The results may be exploited for further design and virtual screening for some novel InhA inhibitors.
(5)Chen, X.-M.; Lu, T.; Lu, S.; Li, H.-F.; Yuan, H.-L.; Ran, T.; Liu, H.-C.; Chen, Y.-D. Structure-Based and Shape-Complemented Pharmacophore Modeling for the Discovery of Novel Checkpoint Kinase 1 Inhibitors. Journal of molecular modeling 2010, 16, 1195–1204.
Checkpoint kinase 1 (Chk1), a member of the serine/threonine kinase family, is an attractive therapeutic target for anticancer combination therapy. A structure-based modeling approach complemented with shape components was pursued to develop a reliable pharmacophore model for ATP-competitive Chk1 inhibitors. Common chemical features of the pharmacophore model were derived by clustering multiple structure-based pharmacophore features from different Chk1-ligand complexes in comparable binding modes. The final model consisted of one hydrogen bond acceptor (HBA), one hydrogen bond donor (HBD), two hydrophobic (HY) features, several excluded volumes and shape constraints. In the validation study, this feature-shape query yielded an enrichment factor of 9.196 and performed fairly well at distinguishing active from inactive compounds, suggesting that the pharmacophore model can serve as a reliable tool for virtual screening to facilitate the discovery of novel Chk1 inhibitors. Besides, these pharmacophore features were assumed to be essential for Chk1 inhibitors, which might be useful for the identification of potential Chk1 inhibitors.
(6)Zhang, Y. L.; Wang, Y. M.; Qiao, Y. J. Combinatorial Screening of COX-2 Inhibitors From Chinese Herbs Based on Multiple Structure-Based Pharmacophores. AMR 2013, 791-793, 269–273.
Ten structure-based pharmacophore models of Cyclooxygenase 2 (COX-2) inhibitors were generated by LigandScout based on COX-2 inhibitor complexes from the Protein Data Bank (PDB). The potential COX-2 inhibitors were identified from traditional Chinese medicine with the method of combinatorial screening with ten models. Based on the screening results of MDDR and the metrics of E, A% and comprehensive appraisal index (CAI), the threshold of hit frequency of mols. was defined and used to identify the active mols. from Chinese herbs. The mols. hit by not less than six pharmacophore models were taken as the screening objects of COX-2 inhibitor and 1103 mols. were obtained.
(7)Zhang, Y. L.; Wang, Y. M.; Qiao, Y. J. Sub-Pharmacophore Generation of JNK3 Inhibitors. AMM 2013, 444-445, 1756–1760.
The structure-based pharmacophore (SBP) model is consisted by the complementarity of the chem. features and space of the interaction between the ligand and receptor. The SBP models always have a high specificity which can only represent the specific class of the ligand. To simplify the models, sub-pharmacophore was then proposed in present study, and was expected to have and only have the most important or the common chem. features which play the major role in the interaction of ligand and receptor. Sub-pharmacophore should contain 4-6 features, the higher specificity with more features, and vice versa. The sub-pharmacophore was generated by the random combination of features from the structure-based models. With the MDL Drug Data Report database used as the testing database, a new metric CAI (comprehensive appraisal index), which integrated the metrics of E and A%, was defined and used to det. the best sub-pharmacophore model. C-Jun N-terminal kinase (JNKs) is one of the mitogen-activated protein kinase family, and widely involved in immune response and inflammatory response, and other pathol. processes. JNK3 is mainly distributed in the brain and nervous system. In present study, twenty-five initial SBP models of JNK3 inhibitors were directly constructed from the Protein Data Bank (PDB) complexes by the LigandScout software. Then, 1018 sub-pharmacophore models were obtained from the 25 initial models. Finally, the best sub-pharmacophore was detd. which was simplified from the initial model generated from the 3FI2 complex, and included four features: one hydrogen bond donor, one hydrogen bond acceptor, and two hydrophobic groups. The metrics of E, A% and CAI value of the best sub-pharmacophore model are 17.47, 31.15 and 5.44, resp. The potential JNK3 inhibitors were then identified from Chinese herbs with the best sub-pharmacophore model, and 286 compds. were obtained.
(8)Zhang, Y. L.; Wang, Y. M.; Qiao, Y. J. Structure-Based Pharmacophore Models Generation and Combinatorial Screening of ICE Inhibitors. AMM 2013, 347-350, 1216–1220.
Eight structure-based pharmacophore models of Interleukin-1β converting enzyme (ICE) inhibitors were generated by LigandScout based on ICE inhibitor complexes from the Protein Data Bank (PDB). A combinatorial screening method based on multiple pharmacophore models were proposed in present study, since the binding pockets of different complexes were different, the structure-based pharmacophore models have a high specificity and cannot cover all the active mols. Based on the screening results of MDDR and the metrics of E and A %, a new metric CAI (comprehensive appraisal index) was defined and used to det. the threshold of hit frequency of mols. which screened by the combinatorial screening method. According to the threshold, the potential ICE inhibitors were then identified from traditional Chinese medicine with the method of combinatorial screening with eight models. The mols. hit by not less than five pharmacophore models were taken as the screening objects of ICE inhibitor, and 781 mols. were obtained.
更多的应用文献,请。
七,联系我们
广州市墨灵格信息科技有限公司
Home Page:
Tel: 020-38261356
Email: info@molcalx.com
Inte:ligand网站:
风险提示:丁香通仅作为第三方平台,为商家信息发布提供平台空间。用户咨询产品时请注意保护个人信息及财产安全,合理判断,谨慎选购商品,商家和用户对交易行为负责。对于医疗器械类产品,请先查证核实企业经营资质和医疗器械产品注册证情况。
 询价记录
询价记录
 技术资料
技术资料
需要更多技术资料 索取更多技术资料
2013_hCAVII_case_study.pdf 附 (下载 20 次)




